Discussion
Alkylating melanotropin fragment analogs
Helga Süli-Vargha
Research Group of Peptide Chemistry, Hungarian Academy of Sciences
H-1518 Budapest 112. P.O. Box 32, Hungary
All knowledge acquired by investigations on biological activities,
structure-activity relationships and mechanism of action of peptide hormones mainly serves
therapeutic purposes. In endocrine disorders the use of good agonists or antagonists is
necessary to mention only the clinical application of insulin, ACTH, TRH, LH-RH and their
analogs, while in other cases the peptide hormones are only carriers of drugs, taking
advantage of their specific receptor recognizing ability.
Whichever may we wish to use peptide hormones in therapeutics, the main requirement is
that the hormone or its analog should possess only one single biological activity so that
it should only bind to the cells of the targeted organ. This aspect has come all the more
into the foreground since in the course of far-reaching investigations on peptide hormones
it turned out that most of them exert various kinds of physiological activities meaning
that they may have various receptors on different cells.
This is the case with the
a-melanocyte stimulating hormone (a-MSH) too, affecting beside its main biological activity (pigment dispersion, skin darkening), the nervous and immune system as well (1). These biological activities constitute the basis of the biomedical application of the a-MSH. In the course of the study of its structure and melanocyte stimulating activity relationships superpotent agonists (2) and antagonists (3) were developed as promising candidates for the treatment of pigment disorders, while among its analogs acting on CNS there are some which are able to improve memory (4). In the therapy of melanomas containing a-MSH receptor positive cells a good cytostatic effect may be expected by using melanotropin antagonists similarly as LH-RH antagonists used in the treatment of prostate cancer (5), or by the application of conjugates containing the hormone and a known cytotoxic agent. Melanotropin antagonits are already available as mentioned above, although to our best knowledge their cytostatic activity has not yet been investigated. As carrier molecule, ß-MSH was used, and its conjugate with Daunomycin (6) showed a selective toxicity in vitro on Cloudman S91 mouse melanoma cells, but there has been no report on in vivo results.In our conception there is no need for the whole hormone molecule for targeting, since it has been known for a long time that smaller fragments of
a-MSH also exert biological activity even if their effect is many orders of magnitude behind that of the parent hormone.The central His-Phe-Arg-Trp sequence can be regarded as the main active center (7), although Eberle et al (8) reported the presence of a second active center the C-terminal Lys-Pro-Val-NH2 sequence, and Medzihradszky and Medzihradszky-Schweiger (9) found other smaller fragments to be active as well in the frog skin bioassay (10). As these investigations were performed in different laboratories and occasionally even on skins of different Rana species, Hruby and coworkers made systematic investigations with peptides uniformly acetylated on their N-terminus and amidated on their C-terminus and their results in the frog skin assay only pointed to the presence of one single centrally located active center. However, their investigations do not preclude the possibility that other melanotropin fragments may be active in another species, or on other than pigment cells, like the Trp-Gly-Lys-Pro-Val-NH2 C-terminal pentapeptide which is active in the tyrosinase assay performed on Cloudman S-90 melanoma cells (12).
Thus, to obtain targeting conjugates, we chose both central and C-terminal fragments of the
a-MSH, like the 5-10 hexapeptide, the 11-13 tripeptide, the 10-13 tetrapeptide, and the 9-13 pentapeptide. The most obvious site for the modification of the peptides with the drug seemed to be the N-terminal part, since the native hormone possesses no free amino terminus, and the biological activity of the smaller fragments also changes favourably with acetylation.As a first approach (13) we substituted the amino terminus of the peptides by the N-(2-chloroethyl)-N-nitrosocarbamoyl (QNO) group, which is the functional group of such effective antitumor agents as BCNU, CCNU and Chlorozotocin. Since in the case of QNO-Glu-His-Phe-Arg-Trp-Gly-OMe we always observed some denitrosation attributable to the presence of the
?-carboxyl group of the glutamic acid, we synthesized an analog of this central sequence where glutamic acid was substituted by glycine and L-phenylalanine by D-phenylalanine to prevent rapid enzymatic degradation of the peptide moiety in the circulation. In this way we got a superactive hexapeptide which was only by one order of magnitude less potent in the frog skin assay than the native tridecapeptide hormone (14).In the other group of conjugates (15) for peptide carriers we chose about the same melanotropin fragments as before and as alkylating agent the phenylalanine mustard (PAM). As compared with the QNO group, we thought that the following characteristics of PAM would be more favourable for our purposes: i) being an amino acid derivative, it can be incorporated into each position of the carrier peptide, ii) the mustard group is more stable than the QNO one and iii) the reaction of the mustard group is unambiguously alkylation, while the chloroethylnitrosourea conjugates have a carbamoylating activity beside the alkylating activity as well.
After synthesizing these melanotropin fragment derivatives the question arose, whether the modified peptides preserved their receptor recognizing ability. Since MSH receptors have been cloned only recently (16), the receptor recognizing ability of the peptide conjugates could only have been checked indirectly, by measuring their biological activity (14). As it can be seen in Table I., the melanocyte stimulating activity of the acetyl and QNO-peptides does not differ significantly, the decrease in the latter case not exceeding one order of magnitude.
The melanotropin fragments containing L- or D-PAM have approximately the same melanin dispersing activity as the parent peptides. In Table I. we have listed only those derivatives which contain the L isomer, Melphalan (Mel), and which were further examined. Since both types of congeners possess biological activity and presumably receptor recognizing ability too, at least in the frog skin bioassay, the basic condition of targeting seems to be fulfilled.
It is well known that some of the
a-MSH analogs and fragments possess prolonged biological activity (17), which means that the skins darkened by the agonist do not lighten to the original colour after washing as the native hormone does.Table I. The melanocyte stimulating activity of a-MSH fragments and
their derivatives
_________________________________________________________________________________
Activity
No. Peptides U/mmol
_________________________________________________________________________________
a-MSH
Ac-Ser-Tyr-Ser-Met-Glu-His-Phe-Arg-Trp-Gly-Lys-Pro-Val-NH2
4x1010
Ac-Lys-Pro-Val-NH2
3x104
I
QNO-Lys-Pro-Val-NH2
6x103
Ac-Gly-Lys-Pro-Val-NH2
5x104
II
QNO-Gly-Lys-Pro-Val-NH2
5x103
Ac-Trp-Gly-Lys-Pro-Val-NH2
6x105
III
QNO-Trp-Gly-Lys-Pro-Val-NH2
4x104 +
Ac-Gly-His-D-Phe-Arg-Trp-Gly-OMe
1x109
IV
QNO-Gly-His-D-Phe-Arg-Trp-Gly-OMe
4x108 +
H-Lys-Pro-Val-NH2
1x104
V
Mel-Lys-Pro-Val-NH2
1x104 +
H-Trp-Gly-Lys-Pro-Val-NH2
7x104 +
VI
Mel-Trp-Gly-Lys-Pro-Val-NH2
2x104 +
VII
Phe-Glu-His-Phe-Arg-Trp-Gly-OMe
2x106 +
VIII
Mel-Glu-His-Phe-Arg-Trp-Gly-OMe
3x106 +
IX
Nle-Glu-His-Mel-Arg-Trp-Gly-OMe
2x106 +
_________________________________________________________________________________
+ Indicates prolonged biological activity.
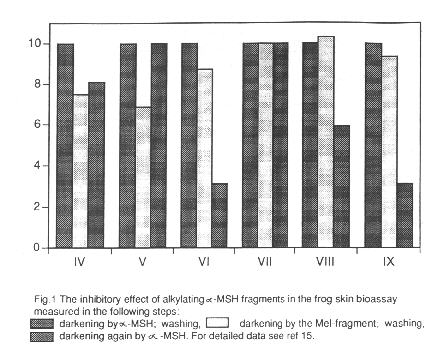
Interestingly not all of the alkylating derivatives cause prolongation of the darkening effect, while some of the normal (unsubstituted) peptides do. For closer investigation of this phenomenon we have performed inhibitory experiments with the peptides marked with +, and the results are presented in Fig. 1. It can be seen that only after the treatment with Phe-Glu-His-Phe-Arg-Trp-Gly-OMe and Mel-Lys-Pro-Val-NH2 could the full darkening effect of the
a-MSH be achieved, while the other derivatives exert inhibitory effect. Accordingly, when Mel replaces methionine, arginine or phenylalanine in the original sequence and in the case of the QNO derivative of the central melanotropin fragment a prolonged action and inhibition occur. Although not proved yet, we think that in these cases a covalent bond is formed between the alkylating group and an appropriate nucleophile on the receptor site to which the Met-Glu-His-Phe-Arg sequence fits. The reason for the long lasting effect of the other peptides, including normal peptides mentioned in the literature without any chemically reactive group, is still unexplained.
The antitumor activity of the alkylating melanotropin fragments has been tested in several in vivo and in vitro systems. The QNO-peptides generally cause a higher increase in the life span of L1210 leukemia bearing mice than the parent antitumor drug BCNU they were nearly equally active on human melanoma xenografts in mice and no effect was observed on human colon xenografts in mice (18). It is interesting that the great differences in the melanocyte stimulating activities (4-5 orders of magnitudes) of the conjugates are not reflected in the melanoma growth inhibitions. The members of the Melphalyl peptide group were effective in the increasing of the life span of L1210 leukemia bearing mice similarly as Melphalan, the only exception was VI with a significant higher effect, and they were roughly equally potent on human melanoma xenografts (19).
The failing of the desired high effect in the case of both types of peptide conjugates on melanoma xenografts does not really contradict the receptor mediated action, the reason for it may be simply the low number of MSH receptors available on the cell surface and, consequently, a relatively low concentration inside of the cell. Moreover it is also known that not all types of human melanoma cells contain
a-MSH receptors (21). Therefore we have chosen several Mel conjugates for further investigations which were carried out by Ghanem and his coworkers at the Free University of Brussels (22). The receptor binding ability of the Mel containing derivatives V, VI, VIII and IX was checked on an a-MSH receptor positive human melanoma cell line against 125J-(Nle4, D-Phe7) a-MSH. Only the central hormone fragment derivatives VIII and IX are competing with the labelled hormone similarly to the natural a-MSH4-10 sequence, and when the cells were preincubated with them, the a-MSH binding was significantly inhibited. It is worth recalling that VIII and IX exerted inhibitory effect on the biological activity in the frog skin bioassay, too (Fig. 1).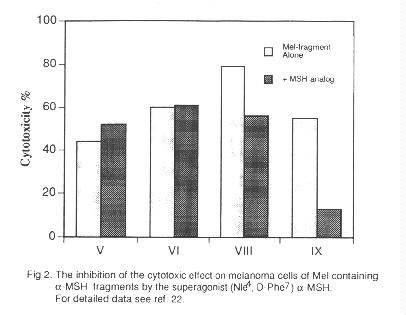
The cytotoxic effect of V, VI, VIII and IX was tested by the 3HTdR uptake assay on human cell lines. The comparison of the drug concentrations inhibiting 50% of cell growth indicates a selective cytotoxic effect on the melanoma cells in the case of VIII and IX which are about six times more toxic on melanoma than on fibroblast cells, while V and VI are equally toxic on both cell lines. When melanoma cells were preincubated with a known
a-MSH superagonist (Nle4, D-Phe7) a-MSH (23) no change in the cytotoxic effect of the C-terminal sequence analogs V and VI occurred, while in the case of the receptor specific central fragment analogs a significant decrease in the activity was observed (Fig. 2), indicating the receptor mediated cytotoxic effect of these congeners.Although not strictly connected with targeting, an advantage of the conjugates should be mentioned, namely their diminished mutagenic effect on normal human lymphocytes as compared to that of Melphalan (20).
Summing up, the synthesis of alkylating analogs of
a-melanotropin fragments, and the investigation of their biological activity may offer answers to some important questions. A number of analogs prove to be irreversible inhibitors, or tightly bound ligands with prolonged biological action. Some conjugates show lower toxicity and better therapeutic indices than the alkylating compound itself. These observations point to the possibility of developing alkylating peptide derivatives advantageously applicable for therapeutic purposes.References
1. Eberle, A.N.: The melanotropins. Basel: Karger, 1988.
2. Chaturvedi, D.N., Hadley, M.E.: The melanotropic peptides Vol. III. p. 129. Ed. M.E.
Hadley, CRC Press, Boca Raton, Florida, 1988.
3. Al-Obeidi, F., Hruby, J.V., Hadley, M.E., Sawyer, T.K., Castrucci, A.M.L.: Int. J.
Peptide Protein Res. 35, 228 (1990).
4. Wied D. de, Jolles, J.: Physiol. Rev. 62, 976 (1982).
5. Schally, A.V., Comaru-Schally, A.M., Redding, T.W.: Proc. Soc. Exp. Biol. Med. 175,
259 (1984).
6. Varga, J.M., Asato, N., Lande, S., Lerner, A.B.: Nature 267, 56 (1977).
7. Otsuka, H., Inouye, K.,: Bull. Chem. Soc. Jpn. 37, 1465 (1964).
8. Eberle, A.N., Schwyzer, R.: Helv. Chim. Acta, 58, 1528 (1975).
9. Medzihradszky, K., Medzihradszky-Schweiger, H.: FEBS Lett. 67, 45 (1976).
10. Shizume, K., Lerner, A.B., Fitzpatrick, T.B.: Endrocrinology 54, 553 (1954).
11. Hruby, V.J., Wilkes, B.C., Hadley, M.E., Al-Obeidi, F., Sawyer, T.K., Staples, D.J.,
Vaux, A.E. de, Dym, O.,Castrucci, A.M.L. de, Hintz, M.F., Riehm, J.P., Rao, K.R.: J. Med.
Chem. 30, 2126 (1987).
12. Eberle, A.N., Kriwaczek, V.M., Schwyzer, R.: Peptides, structural and biological
function. p. 1033. Eds. Gross, E., Meienhofer, J. Pierce Chem. Comp., Rockford IL, 1979.
13. Süli-Vargha, H., Medzihradszky, K.: Int. J. Pept. Protein Res. 23, 650 (1984).
14. Medzihradszky-Schweiger, H., Süli-Vargha, H., Bódi, J., Medzihradszky, K.: Coll.
Czech. Chem. Commun. 53, 2574 (1988).
15. Süli-Vargha, H., Botyánszki, J., Medzihradszky-Schweiger, H., Medzihradszky, K.:
Int. J. Pept. Protein Res. 36, 308 (1990).
16. Mountjoy, K.C., Robbins, L.S., Mortrud, M.T., Coue, R.D.: Science, 257, 1248
(1992).
17. Wilkes, B.C., Sawyer, T.K., Hruby, V.J., Hadley, M.E.: Int. J. Pept. Protein Res. 22,
313 (1983).
18. Jeney, A., Kopper, L., Nagy, F., Lapis, K., Süli-Vargha, H., Medzihradszky, K.:
Cancer Chemother. Pharmacol. 16, 129 (1986).
19. Süli-Vargha, H., Csukás, I., Botyánszki, J., Jeney, A., Kopper, L., Lapis, K.: J.
Cancer Res. Clin. Oncol. 116, 939 (1990).
20. Süli-Vargha, H., Jeney, A., Kopper, L., Oláh, J., Lapis, K., Botyánszki, J.,
Csukás, I., Györvári, B., Medzihradszky, K.: Cancer Lett. 54, 157 (1990).
21. Ghanem, G., Comunale, J., Libert, A., Vercammen, A., Lejeune, F.: Int. J. Cancer 41,
248 (1988).
22. Morandini, R., Süli-Vargha, H., Libert, A., Loir, B., Botyánszki, J., Medzihradszky,
K., Ghanem, G.: Int. J. Cancer, in press.
23. Sawyer, T.K., Sanfilippo, P.J., Hruby, V.J., Engel, M.H., Heward, C.B., Burnett, J.B.,
Hadley, M.E.: Proc. Nat. Acad. Sci. USA 77, 5754 (1980).